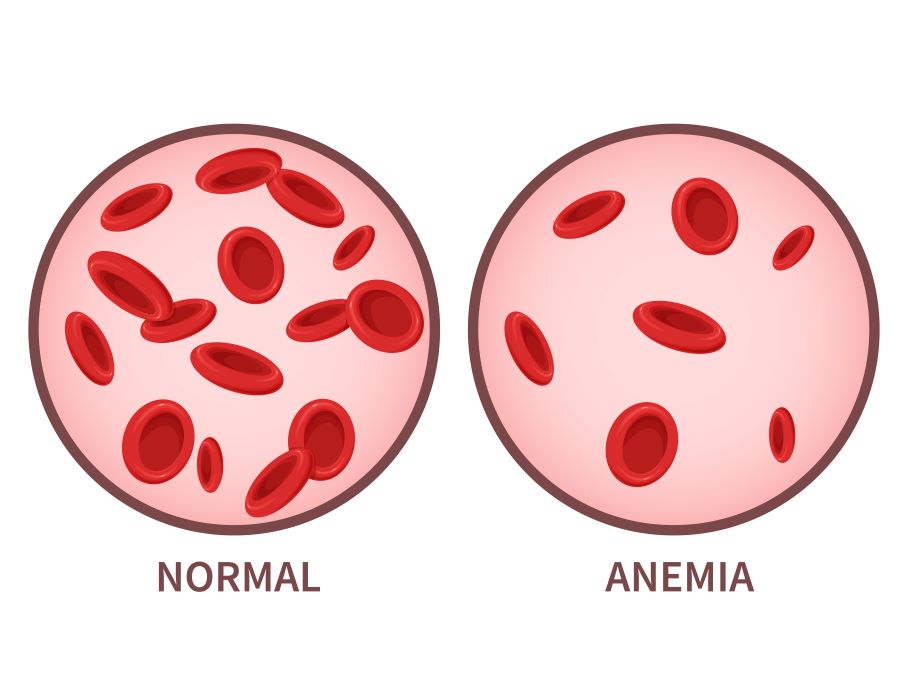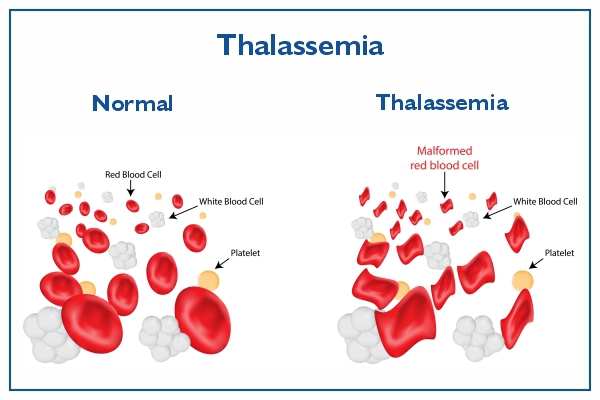

Thalassemia is a genetic blood disorder characterized by abnormal hemoglobin
production, leading to anemia. This condition affects millions of people worldwide,
particularly those of Mediterranean, African, Middle Eastern, and Southeast Asian
descent. Despite being a hereditary disorder, thalassemia varies in severity, with some
individuals experiencing mild symptoms while others require lifelong medical
intervention.
Causes
Thalassemia results from mutations in the genes responsible for hemoglobin
production. Hemoglobin is a protein found in red blood cells that carries oxygen
throughout the body. When these genes are mutated, the production of hemoglobin is
disrupted, leading to insufficient levels of healthy red blood cells.
The severity of thalassemia depends on the specific gene mutations inherited from both
parents. Individuals who inherit one mutated gene are carriers, often asymptomatic but
can pass the condition to their children. Those who inherit two mutated genes, one from
each parent, develop thalassemia.
Types of Thalassemia
- Alpha Thalassemia: This type occurs when there is a defect in the genes
responsible for alpha-globin production. The severity of alpha thalassemia
varies, ranging from mild anemia to a life-threatening condition known as
hydrops fetalis. - Beta Thalassemia: Beta thalassemia results from mutations in the beta-globin
genes. It is further classified into thalassemia major, intermedia, and minor
based on the severity of symptoms.
Symptoms
The signs and symptoms of thalassemia can vary widely depending on its type and
severity. Common symptoms include:
- Fatigue and weakness
- Pale or yellowish skin
- Shortness of breath
- Facial bone deformities (in severe cases)
- Delayed growth and development (in children)
- Enlarged spleen and liver
Severe forms of thalassemia may require regular blood transfusions to alleviate
symptoms and maintain adequate hemoglobin levels. However, repeated transfusions
can lead to iron overload in the body, necessitating treatment with iron-chelating
medications.
Treatment
The treatment approach for thalassemia depends on its type and severity. Options may
include:
Blood Transfusions: Regular transfusions can help replenish red blood cells and
alleviate anemia symptoms.
Iron Chelation Therapy: This treatment removes excess iron from the body, preventing
organ damage caused by iron overload.
Bone Marrow Transplantation: For severe cases, a bone marrow transplant may be
considered to replace defective stem cells with healthy ones.
Folic Acid Supplements: Folic acid supplementation is often recommended to support
red blood cell production.